
ABSTRAK
Nukleosom berfungsi sebagai unit dasar pengemasan kromatin dan memainkan peran penting sebagai hub sentral dalam regulasi epigenetik. Posisi mereka di seluruh genom tidak acak dan mengikuti pola tertentu, dipengaruhi oleh urutan DNA, interaksi histon-DNA, barier fisik kromatin, pergeseran dan pelepasan nukleosom, dan modifikasi kromatin. Ada banyak teknik eksperimental untuk mengidentifikasi posisi nukleosom, tetapi metode ini sering kali melibatkan trade-off antara mencapai resolusi tinggi dan mencakup seluruh genom. Dalam hal ini, pendekatan komputasional dapat menawarkan alternatif cepat, dengan manfaat membantu analisis eksperimental dengan menghilangkan derau data, menyempurnakan batas nukleosom, dan mengidentifikasi fitur yang penting untuk posisi nukleosom. Selain itu, prediksi komputasional memungkinkan integrasi data posisi nukleosom dengan kumpulan data genomik dan epigenomik lainnya, memberikan pandangan yang lebih komprehensif tentang organisasi kromatin dan regulasi gen. Dalam tinjauan ini, kami berfokus pada berbagai metode penentuan posisi nukleosom, termasuk teknik eksperimental identifikasi batas nukleosom dan metode in silico pengurangan derau data penentuan posisi nukleosom dan prediksi penentuan posisi nukleosom dari urutan DNA.
Abstrak Grafis
Alur kerja metode identifikasi dan prediksi untuk posisi nukleosom.
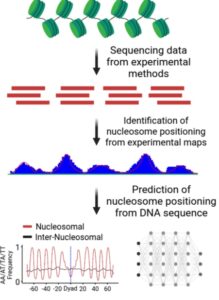
1 Pendahuluan
Nukleosom adalah unit struktural pengemasan kromatin dan hub regulator epigenetik utama. Nukleosom sangat melimpah dalam genom; misalnya, sel haploid manusia mengandung 15 juta nukleosom. Meskipun ini telah menjadi topik perdebatan selama beberapa waktu [ 1 ], banyak penelitian sekarang memberikan bukti kuat bahwa posisi nukleosom tidak acak, khususnya di daerah genom tertentu. Faktanya, nukleosom biasanya berkumpul menjadi susunan, di mana jarak antara nukleosom yang berdekatan tetap relatif konsisten dan bergantung pada organisme [ 2 , 3 ]. Susunan bertahap tersebut selaras dengan baik di hilir situs awal transkripsi gen yang diekspresikan, sedangkan mereka menunjukkan keteraturan rendah pada promotor dan penambah [ 4 ]. Secara keseluruhan, posisi nukleosom mengikuti pola tertentu yang ditentukan oleh faktor-faktor berikut: preferensi urutan DNA, kekuatan interaksi histon-DNA, barier fisik kromatin, pelepasan DNA spontan dari histon, pergeseran nukleosom oleh remodeler yang bergantung pada ATP dan, akhirnya, oleh modifikasi kovalen histon atau DNA [ 5 – 7 ]. Posisi nukleosom dapat didefinisikan berdasarkan dua orientasi. Posisi nukleosom rotasional mengacu pada orientasi DNA di sekitar oktamer histon, yang menggambarkan pasangan basa mana yang menghadap ke arah atau menjauhi inti histon, sedangkan posisi nukleosom translasi mengacu pada lokasi genom nukleosom atau diade nukleosom, di mana diade adalah pusat DNA nukleosom.
Sebagian besar teknik eksperimental yang mengidentifikasi posisi nukleosom membutuhkan banyak sumber daya dan waktu, terutama untuk genom besar seperti manusia. Sejauh ini, penelitian ini telah mengungkap posisi nukleosom untuk sebagian kecil genom tersebut pada resolusi yang cukup tinggi. Di sisi lain, metode komputasi dapat memberikan prediksi cepat di seluruh genom yang juga dapat membantu menghilangkan derau pada data eksperimen, menyempurnakan batas nukleosom, atau menginterpretasikan fitur yang penting untuk posisi nukleosom. Selain itu, memprediksi posisi nukleosom dapat memungkinkan integrasi data posisi nukleosom dengan kumpulan data genomik dan epigenomik lainnya untuk mendapatkan gambaran yang lebih lengkap tentang organisasi kromatin dan regulasi gen. Di sini, kami meninjau berbagai metode posisi nukleosom, baik eksperimental maupun komputasional. Kami menjelaskan beberapa aspek komputasional dari metode tersebut, termasuk identifikasi posisi nukleosom dari peta nukleosom eksperimental dan prediksi posisi nukleosom dari urutan DNA.
2 Metode Eksperimen untuk Pemetaan Posisi Nukleosom
Kami menguraikan beberapa teknik eksperimen utama yang dapat digunakan untuk memetakan posisi nukleosom. Banyak dari metode ini berasal dari teknik DNA-footprinting klasik untuk menyelidiki interaksi protein-DNA, mengingat bahwa nukleosom adalah kompleks histon dengan DNA. Saat ini, teknik ini digunakan bersama dengan teknologi sekuensing generasi berikutnya dan mungkin memerlukan kedalaman sekuensing yang substansial untuk menyediakan pemetaan nukleosom beresolusi tinggi. Misalnya, untuk memetakan posisi nukleosom di seluruh genom pada manusia dengan resolusi setidaknya beberapa pasangan basa, diperlukan sekitar 1–4 miliar pembacaan, dan oleh karena itu, hanya sedikit penelitian yang mencoba memetakan posisi nukleosom di seluruh genom untuk manusia [ 8 – 10 ].
Metode eksperimental yang paling banyak digunakan untuk identifikasi posisi nukleosom di seluruh genom adalah MNase-seq (Gambar 1A ). Metode ini melibatkan pencernaan DNA genom dengan enzim nuklease mikrokokus (MNase), diikuti oleh pengurutan generasi berikutnya [ 11 – 16 ]. Enzim tersebut memiliki aktivitas endonuklease dan eksonuklease, yang memungkinkannya untuk memotong dan merapikan DNA. Dengan kemampuan endonukleasenya, MNase pertama-tama membelah DNA di daerah yang terbuka antara nukleosom atau antara nukleosom dan protein terikat DNA lainnya. Kemudian, aktivitas eksonukleasenya memungkinkannya untuk memotong DNA dari ujung yang terbuka hingga bertemu batas nukleosom atau faktor kromatin. Hasilnya, “jejak” mono-, di-, dan tri-nukleosom diproduksi, bersama dengan banyak fragmen DNA terikat protein [ 17 ]. Setelah melakukan pengurutan generasi berikutnya dari fragmen DNA ini, pembacaan pengurutan yang dihasilkan diselaraskan dengan genom referensi untuk memperoleh lokasi genom fragmen asli. Panjang fragmen DNA yang diperoleh dipengaruhi oleh konsentrasi enzim dan durasi pencernaan. Fragmen pendek, yang dihasilkan melalui pencernaan berlebihan, dapat ditafsirkan secara tidak tepat sebagai daerah bebas nukleosom atau sebagai nukleosom yang telah direnovasi atau tidak terbungkus. Namun, mengekstraksi fragmen dengan panjang sekitar 147 bp, yang sesuai dengan mono-nukleosom, dapat mengurangi masalah ini. Ekstensi utama MNase-seq baru-baru ini mencakup pengurutan nuklease mikrokokus sel tunggal (scMNase-seq), yang mengukur posisi nukleosom dan aksesibilitas kromatin dalam sel tunggal [ 18 ].

GAMBAR 1
Buka di penampil gambar
Presentasi PowerPoint
Ilustrasi metode eksperimen yang dapat mengidentifikasi posisi nukleosom dengan presisi tinggi. (A) Enzim MNase membelah DNA di daerah yang tidak terlindungi. (B) Dengan menggunakan pencernaan DNA berbasis MNase dan antibodi spesifik histon, metode MNase-ChIP-seq dapat mengidentifikasi posisi nukleosom untuk histon tertentu dengan presisi tinggi. (C) Antibodi spesifik histon ditandai dengan enzim MNase untuk mengikat dan memotong di lokasi nukleosom, memungkinkan pemetaan nukleosom yang tepat setelah pemurnian dan pengurutan DNA. (D) Agen khelasi tembaga mengikat histon bermutasi H4S47C dan, setelah penambahan Cu dan H 2 O 2 , menghasilkan radikal hidroksil yang membelah DNA nukleosom di dekat diad, memungkinkan pemetaan posisi nukleosom beresolusi tinggi yang mengapit sumbu diad. HRF: jejak radikal hidroksi; TF: faktor transkripsi.
Protein non-histon yang terikat pada DNA dapat menambahkan sejumlah besar noise pada data MNase-seq, dan oleh karena itu, Chromatin Immunoprecipitation Sequencing (ChIP-Seq) yang dikombinasikan dengan pencernaan berbasis MNase telah diusulkan (MNase-ChIP-seq) [ 19 , 20 ]. Dalam pendekatan ini, antibodi spesifik histon digunakan untuk menargetkan nukleosom, dan pencernaan berbasis MNase diterapkan sebelum imunopresipitasi untuk menghasilkan ukuran fragmen DNA yang diperlukan (Gambar 1B ). Meskipun metode ini memiliki keterbatasan umum MNase-seq, kemampuannya untuk menargetkan protein histon spesifik—seperti varian histon atau modifikasi pasca-translasi histon—membuatnya sangat cocok untuk pemetaan komprehensif posisi nukleosom yang terkait dengan bentuk histon ini [ 21 , 22 ]. Namun, sejumlah besar varian histon pada organisme tingkat tinggi, dikombinasikan dengan kebutuhan akan antibodi berkualitas tinggi, menghadirkan tantangan signifikan untuk menerapkan MNase-ChIP-seq dalam studi posisi nukleosom di seluruh genom. Uji ChIP-exo adalah metode lain di mana pencernaan DNA dilakukan setelah imunopresipitasi, tetapi membutuhkan lebih banyak DNA masukan [ 23 ].
Meskipun ChIP-seq digunakan secara luas untuk mempelajari interaksi protein-DNA, aplikasinya dalam pemetaan nukleosom genom-lebar terbatas. Karena proses sonikasi dan pemotongan acak DNA, resolusinya tidak cukup untuk memetakan posisi nukleosom dengan benar. Untuk mengatasi keterbatasan ini, pendekatan alternatif yang disebut Cleavage Under Targets and Release Using Nuclease (CUT&RUN) diusulkan [ 24 – 26 ]. Secara khusus, antibodi yang ditandai dengan enzim MNase mengikat protein target (misalnya, histon) dan membelah DNA di kedua sisi daerah terikat (Gambar 1C ). Karena hanya fragmen DNA yang terikat pada protein target yang diurutkan, metode CUT&RUN memiliki rasio signal-to-noise yang lebih tinggi dibandingkan dengan ChIP-seq konvensional, yang membutuhkan lebih sedikit bahan masukan. Dengan demikian, kedalaman sekuensing ~10 kali lipat lebih rendah sudah cukup untuk mendapatkan hasil yang sebanding [ 27 , 28 ]. Kerugian yang perlu diperhatikan dari metode ini adalah enzim terkadang dapat membelah DNA yang jauh dari daerah target secara berurutan namun terletak di dekatnya dalam ruang akibat pelipatan kromatin [ 24 ].
Ketepatan pembelahan DNA enzimatik dibatasi oleh halangan sterik dan efek spesifik urutan. Misalnya, enzim MNase mengikat DNA melalui beberapa pasangan basa dan memiliki bias ~30% lebih tinggi terhadap situs target di hulu A dan T daripada G dan C [ 29 ]. Sebaliknya, hydroxyl radical footprinting (HRF) adalah teknik yang ampuh karena radikal hidroksil dapat membelah DNA secara non-spesifik di semua posisi yang terekspos yang tidak terikat oleh protein dan menawarkan resolusi tinggi, hingga nukleotida tunggal, karena ukurannya yang kecil [ 30-32 ]. HRF telah digunakan untuk mengidentifikasi posisi nukleosom dan menentukan situs pengikatan protein yang terikat nukleosom [ 33 ] . HRF sering diterapkan bersama dengan ikatan silang kimia. Yaitu, Ser47 dari histon H4 dapat dimutasi menjadi sistein untuk secara kovalen menempelkan agen khelasi [ 34-36 ]. Agen khelasi tersebut mengkatalisis pembentukan radikal hidroksil nukleolitik untuk membelah DNA. Karena Ser47 terletak secara simetris di kedua salinan H4, lokasi diad dapat ditetapkan dengan resolusi tinggi dari fragmen yang terbelah (Gambar 1D ). Meskipun metode ini dapat memberikan posisi nukleosom yang tepat, kesulitan eksperimental untuk memutasi semua alel H4 diperparah oleh tingginya jumlah salinan gen H4 yang tersebar di berbagai lokus pada organisme tingkat tinggi. Selain itu, residu sistein dalam protein pengikat DNA non-histon dapat berkontribusi pada pembelahan non-target dan menimbulkan gangguan latar belakang dalam data.
Ada juga teknik yang bertujuan untuk menjelaskan aksesibilitas kromatin. Metode berbasis pencernaan enzimatik, DNase-seq [ 37 ] dan ATAC-seq [ 38 ], masing-masing menggunakan DNase I dan transposase Tn5, dapat memberikan informasi tentang aksesibilitas kromatin tetapi menghadapi masalah spesifisitas sekuens yang mirip dengan MNase-seq dan biasanya tidak cocok untuk pemetaan nukleosom resolusi tinggi. Misalnya, sulit untuk secara akurat membedakan posisi nukleosom dari pencernaan berbasis Dnase I karena enzim dapat membelah DNA nukleosom pada oktamer histon di beberapa posisi, menghasilkan fragmen yang jauh lebih pendek daripada jejak nukleosom. Selain itu, metode ini rumit oleh beberapa langkah penanganan sampel dan kerentanannya terhadap variasi teknis selama pencernaan DNA.
Bahasa Indonesia: Ada dua platform web utama yang tersedia daring yang menyediakan informasi tentang posisi nukleosom. Yang pertama, NucPosDB, mencakup tautan ke kumpulan data pemetaan nukleosom eksperimental yang dikurasi secara manual, yang terdiri dari 245 kumpulan data dari 19 spesies dan 39 kumpulan data untuk Homo sapiens , yang terutama dihasilkan menggunakan MNase-seq atau teknik terkait. Ini juga mencakup tautan ke alat untuk mengidentifikasi dan memprediksi posisi nukleosom [ 39 ]. Platform kedua, NucMap 2.0, menawarkan peta posisi nukleosom di seluruh genom untuk sekitar 35 spesies dengan 250 kumpulan data MNase-seq untuk manusia [ 40 ]. Penting untuk dicatat bahwa banyak kumpulan data berfokus pada wilayah genomik, kondisi, atau varian histon tertentu daripada menyediakan pandangan komprehensif tentang posisi nukleosom di seluruh genom.
3 Algoritma Komputasi untuk Identifikasi Posisi Nukleosom dari Peta Eksperimen
Di sini, kami akan fokus pada metode komputasi yang bertujuan mengidentifikasi okupansi nukleosom dan posisi nukleosom serta memperkirakan skor keyakinan mereka dari data eksperimen. Okupansi nukleosom dapat didefinisikan sebagai fraksi sel di mana lokasi tertentu DNA genom ditempati oleh nukleosom. Memperkirakan okupansi nukleosom relatif mudah, karena melibatkan penghitungan jumlah fragmen DNA yang dipetakan ke wilayah genom tertentu. Namun, mengidentifikasi posisi nukleosom rotasi dan translasi biasanya memerlukan penggunaan metode komputasi. Banyak alat komputasi yang sebelumnya dikembangkan untuk menganalisis data posisi nukleosom eksperimental yang diperoleh dari susunan ubin dan pembacaan sekuensing ujung tunggal. Kemajuan teknologi sekuensing generasi berikutnya, terutama sekuensing ujung berpasangan, saat ini memungkinkan inferensi posisi nukleosom yang lebih akurat, dengan mempertimbangkan fragmen yang lebih panjang dan kedalaman sekuensing yang tinggi. Namun, tantangan terbesar adalah mendeteksi nukleosom fuzzy yang menempati beberapa posisi. Posisi nukleosom dinamis ini muncul dari heterogenitas dalam populasi sel, yang dipengaruhi oleh berbagai variabel, termasuk keadaan epigenetik, kondisi seluler, faktor in vivo, dan perubahan dalam ekspresi gen (Gambar 2 ).
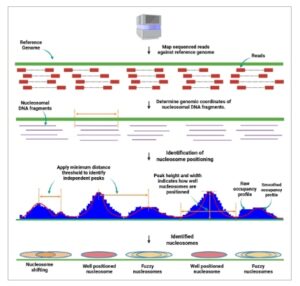
GAMBAR 2
Buka di penampil gambar
Presentasi PowerPoint
Ilustrasi alur kerja umum untuk mengidentifikasi posisi nukleosom dari data MNase-seq eksperimental. Lokasi genomik fragmen DNA ditentukan dengan memetakan pembacaan MNase-seq ke genom referensi. Fragmen yang kira-kira berukuran nukleosom dipilih untuk menghitung hunian nukleosom mentah. Metode komputasional menerapkan teknik denoising untuk mengurangi noise dari profil hunian. Terakhir, amplitudo, lebar, dan jarak nupeak yang teridentifikasi memberikan informasi yang berguna tentang posisi nukleosom.
Karena banyak alat yang dikembangkan sebelumnya kini tidak dapat diakses lagi, berikut ini kami soroti beberapa metode yang tersedia dalam kategori ini (Tabel 1 ). Metode-metode ini menerapkan berbagai teknik penghalusan (konvolusi Gaussian, kernel tri-weight, Fast Fourier Transform) untuk mengurangi noise dan fluktuasi dalam data eksperimen, baik dengan menghaluskan jumlah dyad maupun profil hunian nukleosom [ 41 , 43 , 46 ]. Sebagian besar metode menyaring pembacaan sekuens untuk hanya menyertakan yang sesuai dengan footprint nukleosomal dengan fokus pada pembacaan ~147 bp, memperhitungkan bias yang diperkenalkan oleh pencernaan MNase, dan memberikan ukuran ketidakjelasan, yang menunjukkan variabilitas dalam posisi nukleosom. Keakuratan metode-metode ini biasanya dinilai dengan memulihkan posisi nukleosom dari data eksperimen atau simulasi dan dengan kualitas profil hunian nukleosom dari nukleosom berfase baik pada daerah genomik tertentu [ 12 , 36 , 47 , 48 ].
TABEL 1. Daftar alat komputasi yang mengidentifikasi posisi nukleosom dari data eksperimen.

Catatan: Dengan menekan tombol Ctrl dan mengklik nama alat, Anda akan diarahkan ke halaman aksesnya jika tersedia. Metode-metode ini telah dicantumkan berdasarkan apakah kode atau alatnya tersedia untuk umum.
Singkatan: CLI: antarmuka baris perintah; FFT: transformasi fourier cepat.
Salah satu alat yang paling banyak digunakan dalam kategori ini adalah nucleR, paket R/Bioconductor yang menawarkan metode non-parametrik untuk mengidentifikasi posisi nukleosom dari data MNase-seq dan Tiling Array (TA) [ 41 ]. Alat ini dapat menggunakan pembacaan ujung tunggal atau ujung berpasangan sebagai input, dapat mengoreksi bias untai untuk pembacaan ujung tunggal, dan melakukan pemangkasan pembacaan untuk pembacaan ujung berpasangan. Selanjutnya, menggunakan Fast Fourier Transform (FFT), ia menghilangkan noise dari data, mempertahankan sejumlah kecil komponen (~1%–2%), dan menemukan maksima lokal (puncak) yang sesuai dengan diade nukleosom. nucleR kemudian diadopsi sebagai pemanggil puncak nukleosom dalam rangkaian Dinamika Nukleosom [ 44 ]. Alat lain yang umum digunakan adalah DANPOS [ 42 ]. Ia menghilangkan noise dari data dan melakukan penyesuaian ukuran fragmen. Fitur penting dari metode ini adalah kapasitasnya untuk melakukan analisis diferensial antara sampel dan mengidentifikasi apakah nukleosom diposisikan dengan baik, menunjukkan ketidakjelasan, atau mengalami pergeseran dinamis. Teknik konvolusi Gaussian diterapkan pada profil hunian nukleosom dalam kasus metode iNPS lainnya [ 43 , 49 ]. Tidak seperti metode lain, iNPS menghitung turunan pertama, kedua, dan ketiga dari profil yang dihaluskan untuk menemukan titik belok untuk menemukan puncak dan batas nukleosom. Algoritme ini juga dapat mendeteksi bahu di dekat puncak utama yang sesuai dengan nukleosom fuzzy. Alat ini mengasumsikan bahwa setiap pembacaan dari data MNase-seq sesuai dengan fragmen nukleosom. Namun, diketahui bahwa, dalam kasus MNase-seq, beberapa fragmen dapat dilindungi oleh protein non-histon yang terikat DNA. iNPS dan DANPOS keduanya dapat mendeteksi diad nukleosom dan batas nukleosom.
4 Identifikasi Fitur Posisi Nukleosom
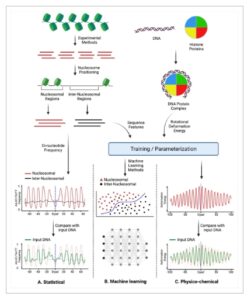
Sementara berbagai faktor memengaruhi penempatan nukleosom dalam genom, sifat DNA intrinsik memainkan peran penting dalam posisi nukleosom, dan kontribusi urutan DNA dalam menjelaskan posisi nukleosom in vitro dan in vivo telah diusulkan menjadi substansial [ 50 , 51 ]. Memang, telah disarankan bahwa biaya deformasi pembengkokan DNA di sekitar oktamer histon mungkin bergantung pada posisi di-nukleotida pirimidin–purin spesifik yang lebih mudah dideformasi daripada motif lain [ 52 – 54 ]. Misalnya, pola urutan di-nukleotida telah dijelaskan di mana motif urutan kaya A/T atau kaya G/C memiliki periodisitas sekitar 10–11 pasangan basa dalam DNA yang terikat nukleosom [ 55 – 58 ]. Pola-pola ini biasanya diringkas sebagai dinukleotida WW (W mewakili A/T) yang diperkaya dalam alur minor DNA nukleosomal yang membengkok ke dalam dan menghadap oktamer histon (ke dalam), dan dinukleotida SS (S mewakili C/G) yang sering ditemukan dalam alur minor yang menghadap ke luar atau, dengan kata lain, alur mayor yang membengkok ke dalam (Gambar 3A ). Pembengkokan alur minor DNA nukleosomal dimediasi oleh penyisipan rantai samping arginin ke dalam alur minor di 14 situs interaksi utama [ 59 – 64 ]. Misalnya, analisis urutan DNA 601 yang diposisikan dengan baik, yang secara khusus dirancang untuk mengoptimalkan dan menstabilkan posisi nukleosom, menunjukkan terjadinya gugus GC yang secara berkala bergantian dengan gugus AT [ 53 ].
GAMBAR 3
Buka di penampil gambar
Presentasi PowerPoint
Representasi konseptual dari strategi komputasional utama untuk memprediksi posisi nukleosom dari urutan DNA. Nukleosom dan daerah bebas nukleosom (antarnukleosom) dapat diidentifikasi dari metode eksperimen. (A) DNA nukleosom biasanya menunjukkan pola periodik ~ 10–11 bp dalam frekuensi nukleotida. Sebagai contoh, dinukleotida AA/AT/TA/TT menunjukkan pola periodik sehubungan dengan oktamer histon dan diad. Metode statistik menilai kemungkinan bahwa urutan DNA tertentu (“DNA masukan”) berasal dari daerah nukleosom berdasarkan seberapa baik urutan tersebut sesuai dengan pola nukleotida karakteristik. (B) Selain pola nukleotida, urutan dan fitur struktural lainnya dapat diekstraksi dan digunakan untuk melatih model pembelajaran mesin untuk membedakan urutan nukleosom dari non-nukleosom. (C) Sifat fisikokimia, seperti energi deformasi yang berasal dari parameter translasi dan sudut DNA, dapat digunakan untuk memprediksi kecenderungan DNA untuk membungkus oktamer histon.
Sementara pola di-nukleotida telah diselidiki di masa lalu, banyak model posisi nukleosom sebelumnya kurang presisi karena kelenturan di-nukleotida bergantung pada konteks sekuens lokal, dan sifat fisikokimia pola sekuens yang lebih panjang tidak bersifat aditif terhadap langkah pasangan basa yang sesuai [ 65 , 66 ]. Baru-baru ini, semakin banyak bukti yang menunjukkan bahwa motif DNA yang lebih panjang juga memainkan peran penting dalam posisi nukleosom. Misalnya, ditunjukkan bahwa ada hubungan yang signifikan antara lokasi pola tri- dan bahkan tetra-nukleotida tertentu dan atribut struktural nukleosom, terutama dalam alur DNA minor dan mayor [ 67 , 68 ]. Selain itu, sekuens poli(dA:dT) juga dianggap sebagai fitur penting untuk posisi nukleosom [ 69 ]. Sekuens ini menunjukkan kekakuan diferensial tergantung pada panjangnya, yang pada gilirannya, memengaruhi inklusi preferensialnya ke dalam daerah terikat nukleosom atau daerah yang kekurangan nukleosom. Selain itu, penelitian sebelumnya menunjukkan bahwa pengaruh pola sekuens dan konten sekuens melampaui batas nukleosom [ 68 , 70 ]. Memang, pengayaan motif A/T yang relatif pendek di daerah penghubung DNA nukleosom mendukung fleksibilitas DNA yang diperlukan untuk mengikat histon H1, dan berkontribusi pada pembentukan struktur seperti batang [ 71 , 72 ].
Meskipun urutan DNA memberikan kontribusi yang signifikan terhadap posisi nukleosom, kemajuan dalam pemodelan komputasional, dikombinasikan dengan teknik eksperimental yang lebih baik, telah menunjukkan bahwa urutan DNA saja tidak dapat sepenuhnya menjelaskan posisi nukleosom. Ada “penghalang” dalam kromatin, yang secara sterik mencegah hunian nukleosom di daerah tertentu [ 73 – 75 ]. Memang, nukleosom disejajarkan pada interval reguler di sekitar situs awal transkripsi (TSS) dan situs pengikatan CTCF [ 76 ]. Selain itu, pengaruh perombak kromatin, seperti kompleks SWI/SNF, yang secara aktif memposisikan ulang atau mengusir nukleosom, menambah lapisan kompleksitas lain untuk mengidentifikasi posisi nukleosom [ 77 ].
5 Strategi Komputasi untuk Memprediksi Posisi Nukleosom dari Urutan DNA
Metode eksperimental untuk menentukan posisi nukleosom memerlukan cakupan kedalaman sekuens yang luas dan dapat mencapai pemetaan nukleosom di seluruh genom untuk organisme tertentu, seperti ragi [ 78 , 79 ], tetapi tampaknya bermasalah untuk genom yang lebih besar, seperti manusia. Oleh karena itu, berbagai metode komputasi telah dikembangkan untuk mengisi celah ini. Kami telah mengkategorikan metode ini ke dalam beberapa kelompok berdasarkan algoritma yang mereka gunakan, meskipun perbedaan antara kategori sering tidak jelas (Tabel 2 , Gambar 3 ).
TABEL 2. Daftar metode untuk memprediksi posisi nukleosom dari urutan DNA.
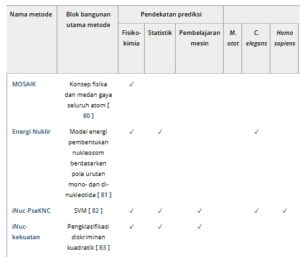
Catatan: Dengan menekan tombol Ctrl dan mengklik nama metode, Anda akan diarahkan ke halaman aksesnya jika tersedia. Metode-metode ini telah dicantumkan berdasarkan apakah kode atau alatnya tersedia untuk umum.
Singkatan: CNN: jaringan saraf konvolusional; SVM: mesin vektor pendukung.
Kelompok pertama metode prediksi posisi nukleosom (Gambar 3A ) didasarkan pada analisis statistik frekuensi motif sekuens DNA, yang mendukung posisi nukleosom, yang telah ditinjau di bagian sebelumnya. Biasanya, sebagai langkah pertama, matriks frekuensi/bobot untuk motif sekuens (kebanyakan di-nukleotida digunakan) pada setiap posisi sekuens nukleosom diperoleh dari penyelarasan fragmen nukleosom eksperimental. Kemudian, posisi nukleosom ditentukan dengan menilai kesamaan antara sekuens DNA yang diberikan dan matriks bobot spesifik posisi yang telah dihitung sebelumnya, dan daerah nukleosom dan posisi dyad diprediksi dengan mengidentifikasi nilai maksimum dari skor kesamaan yang sesuai. Metode selanjutnya berusaha untuk menggabungkan daerah antar-nukleosom dan memperhitungkan pengaruh komposisi sekuens [ 69 , 70 , 87 , 92 , 93 ]. Misalnya, metode RECON membagi DNA menjadi fragmen pendek yang tidak tumpang tindih, menghitung distribusi di-nukleotida untuk setiap fragmen, dan melakukan analisis diskriminan [ 85 ].
Kelompok metode yang dijelaskan sebelumnya biasanya tidak menggunakan pelatihan eksplisit pada dataset nukleosomal dan inter-nukleosomal, tetapi beberapa optimasi parameter masih diperlukan. Teknik pembelajaran mesin yang diawasi mewakili kelompok metode kedua untuk memprediksi posisi nukleosom (Gambar 3B ). Metode klasifikasi ini dilatih pada set positif (nukleosomal) dan negatif (daerah inter-nukleosomal) yang berasal dari data eksperimen. Urutan DNA nukleosomal dan inter-nukleosomal biasanya direpresentasikan sebagai vektor biner yang mengkode motif urutan (dibahas di bagian sebelumnya) dan berbagai fitur fisikokimia [ 82 , 94 ]. Salah satu metode populer, NuPoP, didasarkan pada Model Markov Tersembunyi durasi dan secara eksplisit memodelkan distribusi panjang inter-nukleosomal [ 95 ]. Faktor ini penting, karena panjang pengulangan inter-nukleosomal secara langsung memengaruhi kepadatan nukleosom keseluruhan dalam serat kromatin dalam organisme tertentu [ 96 ]. Analisis perbandingan komprehensif dari metode pembelajaran mesin telah dilakukan sebelumnya, dan menunjukkan bahwa kinerja masing-masing metode bervariasi tergantung pada wilayah genom yang digunakan dalam set pengujian [ 97 ] dan pada kualitas data set pelatihan [ 98 ]. Selain itu, beberapa metode bekerja dengan baik secara konsisten pada ragi, tetapi kinerjanya memburuk ketika diterapkan pada manusia [ 93 , 99 ].
Kemajuan dalam teknik eksperimental, menghasilkan set data genom-lebar berkualitas tinggi yang dapat digunakan untuk pelatihan, telah mengilhami penerapan metode pembelajaran mendalam untuk prediksi NP. Keuntungan dari jaringan saraf adalah kemampuannya untuk menangkap pola kompleks dan hubungan non-linier antara urutan DNA dan posisi nukleosom. LeNup adalah metode pembelajaran mendalam untuk mengklasifikasikan DNA nukleosomal dari inter-nukleosomal; ia menggabungkan konsep jaringan saraf konvolusional, modul awal, dan mekanisme gating [ 91 ]. Metode lain, NuPoSe, menggunakan DNA nukleosomal, daerah penghubung DNA pengapit nukleosom, dan daerah inter-nukleosomal dalam pelatihan model prediksi pembelajaran mendalam [ 68 ]. Selain itu, ia menerapkan pendekatan pemilihan fitur dua langkah yang memungkinkannya untuk mengidentifikasi dan menginterpretasikan 43 fitur yang memainkan peran kunci dalam klasifikasi. Perlu disebutkan bahwa keterbatasan banyak metode pembelajaran mendalam berasal dari kurangnya interpretabilitas dan ketidakmampuan mengidentifikasi fitur penting untuk membentuk nukleosom.
Kelompok ketiga metode prediksi posisi nukleosom (Gambar 3C ) bergantung pada gagasan bahwa molekul DNA dengan energi deformasi rendah memfasilitasi pembungkusan DNA di sekitar oktamer histon [ 93 , 100-106 ]. Energi deformasi DNA bergantung pada urutannya dan mengacu pada jumlah energi yang diperlukan untuk membengkokkan, memelintir , atau merusak molekul DNA dari keadaan rileksnya. Berbagai metode telah dikembangkan yang memperkirakan parameter kekakuan DNA dari kovariansi berpasangan parameter langkah dinukleotida (Roll, Tilt, Twist, dan lainnya) yang diamati dalam struktur kompleks protein-DNA yang diketahui [ 107 ] atau oligonukleotida [ 108 ]. Parameter ini kemudian digunakan untuk menilai kemudahan deformasi urutan DNA yang berbeda pada permukaan oktamer histon dari kelengkungan alaminya, untuk panjang fragmen tertentu. Nilai minimum dalam profil energi deformasi biasanya diambil sebagai posisi nukleosom. Selain itu, beberapa model mempertimbangkan kekuatan interaksi antara histon dan DNA [ 109 ]. Namun, semua metode ini biasanya dikembangkan, dan parameter telah dievaluasi menggunakan sejumlah kecil struktur, dan kinerja prediksi NP genomiknya masih belum jelas.
Secara keseluruhan, metode yang ada untuk memprediksi posisi nukleosom dari urutan DNA memiliki beberapa keterbatasan. Pertama, mayoritas alat yang dijelaskan di atas tidak dapat diakses, atau versi mandiri mereka tidak kompatibel dengan bahasa pemrograman atau pustaka yang berbeda. Kedua, karena kompleksitas komputasi yang tinggi, sebagian besar metode telah dikembangkan berdasarkan data volume rendah yang digunakan untuk pelatihan atau parameterisasi. Selain itu, beberapa metode telah dilatih pada data eksperimen beresolusi rendah, yang menghasilkan akurasi rendah atau prediksi posisi nukleosom acak ketika diterapkan di seluruh genom. Selain itu, nukleosom yang diposisikan dengan baik, yang sering digunakan untuk pelatihan, terletak di daerah genom tertentu, yang dapat menimbulkan bias tambahan. Akhirnya, sebagian besar metode yang disebutkan dapat memprediksi posisi rotasi (memprediksi bagian DNA mana yang menghadap histon atau ke luar tanpa memprediksi lokasi diad) atau membedakan daerah genom nukleosomal dari internukleosomal. Namun, kemampuan mereka untuk memprediksi posisi nukleosom translasi dan, oleh karena itu, lokasi genom diad nukleosom masih terbatas.
6. Penempatan Nukleosom dan Deteksi Dini Penyakit
Prediksi posisi nukleosom dapat membantu menguraikan bagaimana gen dihidupkan atau dimatikan, memfasilitasi diagnostik penyakit. Memahami posisi nukleosom juga dapat membantu dalam mengembangkan obat yang menargetkan struktur kromatin atau modifikasi epigenetik, yang berpotensi meningkatkan pengobatan untuk kelainan genetik dan epigenetik. Misalnya, biopsi cair adalah alat diagnostik baru yang mengidentifikasi materi terkait tumor, termasuk DNA tumor yang bersirkulasi (ctDNA), protein, dan antibodi dalam darah. Ini non-invasif dan memungkinkan karakterisasi komprehensif heterogenitas tumor dan evolusi klonal. Meskipun demikian, ada tantangan dalam membedakan ctDNA dari DNA bebas sel (cfDNA). Telah diketahui bahwa pola fragmentasi cfDNA bersifat non-acak, dengan fragmen yang lebih panjang sesuai dengan DNA nukleosomal [ 110 , 111 ]. Selain itu, distribusi ukuran fragmen dan posisi nukleosom dapat berbeda antara individu yang sehat dan pasien kanker [ 112 , 113 ]. Menentukan posisi nukleosom secara eksperimental untuk setiap sampel biopsi cair merupakan proses yang memakan waktu dan biaya. Akibatnya, hanya sebagian kecil genom yang dapat dipetakan pada cakupan tinggi, dan banyak sampel biopsi cair mungkin tetap tidak terkarakterisasi. Untuk mengisi celah ini, metode in silico berperan untuk menganalisis, menafsirkan, dan mengklasifikasikan pola ctDNA yang khas. Beberapa upaya komputasi telah dikembangkan untuk menggunakan berbagai fitur cfDNA dan ctDNA untuk meningkatkan akurasi deteksi dan klasifikasi fenotipe tumor. Fitur-fitur ini meliputi pola metilasi DNA, distribusi ukuran fragmen DNA, aksesibilitas kromatin, lokasi genom, dan, di antaranya, posisi nukleosom [ 111 , 114 , 115 ].
Dalam studi terbaru, pemetaan nukleosom dari tumor berpasangan dan jaringan normal dari pasien kanker payudara yang sama diproduksi dengan menggunakan histone H3 ChIP-seq berbantuan MNase dan dibandingkan dengan cfDNA dari plasma darah [ 116 ]. Analisis ini mengungkap reposisi nukleosom di beberapa daerah regulasi penting dan perbedaan nukleosom spesifik kanker di antara pasien. Menariknya, hasil menunjukkan bahwa jaringan tumor memiliki panjang pengulangan nukleosom yang lebih pendek daripada jaringan normal, dan skor nukleosom yang tinggi dapat memprediksi keberadaan tumor ganas. Deviasi genom-lebar dari jejak nukleosom cfDNA antara kanker dan jaringan normal berhasil digunakan dalam studi lain untuk mengidentifikasi keberadaan tumor ganas [ 117 ]. Akhirnya, hubungan potensial antara posisi nukleosom dan regulasi penyambungan alternatif dalam sel kanker payudara setelah stimulasi progesteron juga diamati dan diterapkan untuk diagnostik [ 118 ].
Meskipun upaya tak ternilai yang disebutkan di atas dalam deteksi dini kanker berdasarkan cfDNA dan posisi nukleosom, bidang ini menghadapi tantangan signifikan yang menghambat efektivitasnya. Pertama, posisi nukleosom adalah proses dinamis yang dipengaruhi oleh berbagai faktor, termasuk jenis sel, penyakit, dan modifikasi epigenetik. Oleh karena itu, tidak ada profil hunian nukleosom unik yang dapat berfungsi sebagai referensi dalam analisis deteksi penyakit dini. Kedua, cfDNA dilepaskan dari jaringan yang berbeda ke dalam aliran darah, dan saat ini tidak ada metode untuk menentukan jaringan spesifik tempat fragmen cfDNA berasal. Ketiga, karena bidang ini masih berkembang, alat dan teknik tambahan diperlukan untuk mengintegrasikan biologi nukleosom dengan berbagai modifikasi epigenetik untuk deteksi dini dan pemantauan penyakit yang kuat.
7 Diskusi
Di luar aplikasi klinisnya, data posisi nukleosom sangat penting untuk memahami proses biologis fundamental, mengungkap bagaimana susunan nukleosom terlipat menjadi kromatin, bagaimana modifikasi kovalen DNA dan histon mengatur ekspresi gen dan pemadatan kromatin, dan bagaimana pergeseran nukleosom atau pelepasan DNA dari histon memengaruhi pengikatan faktor transkripsi [ 93 , 119 – 122 ]. Namun, posisi nukleosom bersifat statistik, dan setiap pola sekuens dapat ditemukan dalam sejumlah kecil sekuens genom dan sering kali sulit dibedakan dari noise (yang dihasilkan oleh kesalahan eksperimental atau ketidakjelasan nukleosom di sepanjang genom) [ 62 ]. Selain itu, (re-)posisi nukleosom diatur tidak hanya oleh preferensi sekuens tetapi juga sangat dipengaruhi oleh faktor in vivo, kompleks remodeling kromatin, dan lingkungan kromatin lokal, termasuk arsitektur gen [ 123 – 125 ]. Selain itu, pola posisi nukleosom bersifat spesifik terhadap konteks dan mungkin berbeda di berbagai wilayah genom dan organisme [ 21 , 126 ]. Untuk mengatasi masalah ini, para peneliti saat ini mencoba untuk fokus pada model yang lebih disesuaikan untuk memprediksi posisi nukleosom dalam konteks biologis yang beragam [ 127 ].
8 Kesimpulan
Topik identifikasi posisi nukleosom telah mengalami periode antusiasme yang besar dan skeptisisme berikutnya. Sulit untuk menentukan dengan tepat di mana posisi kita saat ini, tetapi jelas bahwa bidang ini menghadapi keputusan penting tentang arah masa depannya dan pertanyaan penelitian yang perlu diprioritaskan. Kita berada di persimpangan jalan, dengan potensi untuk memanfaatkan kemajuan terbaru dalam pemetaan nukleosom genom utuh di samping metode komputasi mutakhir, seperti pembelajaran mendalam. Tantangannya terletak pada menentukan cara terbaik untuk menggunakannya untuk pelatihan model dan wawasan prediktif.